Physical Address
304 North Cardinal St.
Dorchester Center, MA 02124
Physical Address
304 North Cardinal St.
Dorchester Center, MA 02124


We’ve been digging into biodata infrastructure for drug discovery. Here’s what we’ve found so far, what we’re building toward, and what we still can’t figure out.
A build-in-public note from Ridio. We’re a small team exploring a startup idea at the intersection of biological data and AI. This is us sharing our homework, not a pitch deck.
A few weeks ago, we started looking into AI for drug discovery. We didn’t have a product idea yet. We just wanted to understand the space, so we started talking to founders and researchers who are already building in it.
The more conversations we had, the more we kept hearing the same thing. Working with biological data is really, genuinely difficult. Not in the “big data is hard” generic sense, but in a very specific, practical way. The data you need is spread across multiple public databases. Each one uses different formats. The same protein or molecule can have a completely different name depending on where you look it up. And the connections between data points, the relationships that actually matter for drug discovery, aren’t laid out in a way that AI can follow.
What surprised us most was that every team seemed to be solving this problem on their own. Different startups, different pharma teams, different research labs, all writing their own code to pull, clean, and stitch together the same underlying data. There was no common infrastructure to lean on.
That’s when the thesis started to form: better bio-AI might start with better data, not better models. If the data layer is this consistently painful across the industry, maybe the opportunity isn’t building another AI tool on top. Maybe it’s fixing the layer underneath.
Let me be concrete, because “data is messy” is too vague to be useful.
Much of the biological data lives in public databases. These are genuinely amazing resources, decades of scientific work cataloged and made freely available. One database has protein sequences. Another has 3D molecular structures. Another tracks which drug-like molecules interact with which biological targets.
The databases themselves are great. The problem is what happens when you try to use more than one at the same time.
Here’s a real example. Say you’re working on insulin. You want to know what the protein does (that’s in UniProt, where it’s called “P01308”). You want to see its 3D shape (that’s in the Protein Data Bank, where it’s called “4EY1”). You want to know which drug compounds interact with it (that’s in ChEMBL, where it’s called “CHEMBL5881”). And maybe you want clinical variant data too (that’s in ClinVar, where it’s “3630”).
Same protein. Four databases. Four different ID codes. Four different data formats (XML, mmCIF, JSON, VCF). And no built-in way to tell a computer that these four entries are all talking about the same thing.
We’ve boiled the problem down to four specific issues:
The data is scattered. No single database has the complete picture of any one target. You always need multiple sources.
The formats are all different. Every database has its own way of structuring and delivering data. You can’t just merge them.
The naming is inconsistent. The same protein, gene, or molecule gets a different identifier in every database. Reconciling these manually is painful and error-prone.
The relationships are hidden. Even after you’ve solved the first three problems, the connections between data points (which protein binds to which compound, which structure corresponds to which sequence) aren’t spelled out in a way AI can follow. It’s like having all the puzzle pieces but no picture on the box.
These four problems feed into each other. And the practical result is that every team building AI for biology burns a huge amount of time and money on data plumbing before they can build anything useful.
Drug discovery has two big phases.
First is Discovery: figuring out what biological target to go after, finding molecules that might work against it, and narrowing those down to the best candidates.
Then comes Development: testing those candidates in animals and humans, running clinical trials, and getting regulatory approval.
We’re focused on Discovery, specifically the early stages: target discovery (finding what to aim at), hit generation (screening for molecules that interact with the target), and lead identification (picking the most promising ones from the screen).
Why these stages? Because each one requires pulling from multiple databases and reasoning across them. To discover a target, you might need protein function data alongside structural data. To generate hits, you need to cross-reference molecular shapes with known binding data. To identify leads, you need all of the above combined.
This is where the fragmentation hurts most. Later Development stages use different kinds of data entirely (patient records, trial results), and those have their own infrastructure challenges. But in early Discovery, the pain comes from biological databases not talking to each other. That’s the gap we’re looking at.
We need to start somewhere specific. We chose three databases: UniProt, ChEMBL, and the Protein Data Bank (PDB).
Here’s why these three and not others.
UniProt is the starting point for almost any protein-related question. It catalogs what proteins are, what they do, which organisms they appear in. It’s comprehensive and well-maintained. If you’re asking “what does this protein do?”, this is where you go.
PDB stores the 3D shapes of proteins. These are experimentally measured structures that show, at the atomic level, what a protein looks like and where on its surface a drug molecule might attach. If you’re asking “what does this protein look like?”, PDB has the answer.
ChEMBL tracks drug-like molecules and how they interact with biological targets. It records which compounds have been tested, against what, and how potent they are. If you’re asking “what compounds are known to affect this protein?”, ChEMBL is the source.
Together, these three cover the data types you need for structure-based drug design, which is one of the most common approaches in modern drug discovery. And they share something important: UniProt’s accession number (a unique code assigned to each protein) is referenced by both PDB and ChEMBL. That shared ID acts like a thread that ties all three databases together.
In theory, you can follow that thread from “here’s a protein” to “here’s its shape” to “here are known compounds that interact with it.” In practice, making that journey smooth and automated is the hard part, and that’s what we want to build.
We also chose three databases rather than one for a specific reason: the cross-database problem is invisible if you only look at one source. UniProt on its own is beautifully organized. The mess only shows up when you try to combine it with PDB and ChEMBL. Three is the minimum to demonstrate the real problem, and the real value of solving it.
To be clear about what we’re not building: we’re not building a drug discovery AI model, and we’re not trying to replace these databases.
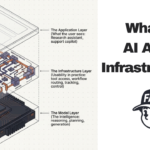
What we’re exploring is a layer that sits between the raw databases and the AI systems that need their data. Think of it as a translator. The underlying content stays where it is. Our layer would make it readable and usable for AI, in a consistent way, across all three sources at once.
Concretely, for the scattering problem, it would give teams a single API instead of three separate integrations. For the format problem, it would deliver all data in one consistent structure regardless of what’s happening underneath. For the naming problem, it would automatically map identifiers across databases so you don’t have to reconcile them yourself. For the hidden connections problem, it would make relationships between proteins, structures, and compounds explicit and navigable.
It would also handle the operational stuff that every team currently deals with on their own: caching frequently requested data, respecting rate limits, and tracking upstream changes when databases update their formats.
The core pitch to any potential user would be simple: focus on your product, not on plumbing.
Bio-AI startups are small teams building specific tools for parts of the drug discovery process. They’re usually technically strong but short on time and money. Right now, they spend their first months building data pipelines before they can start on their actual product. That’s plumbing, not product. We’d let them skip it.
Pharma and biotech companies are deploying AI across their entire discovery workflow, often with many teams working in parallel. Their problem is that dozens of internal teams are independently building overlapping pipelines to the same databases. A shared infrastructure layer would reduce the duplication and make the data consistent.
I want to be honest about what we don’t know, because the list is long and the items are real.
Should we store our own copy of the data, or translate on the fly? If we call the original databases in real time and clean the data as it passes through, we’re lighter to build but we depend on their speed, availability, and rate limits. If we download and store our own cleaned copy, we have more control but we have to keep it in sync as the originals update. The right answer is probably a hybrid, but we haven’t nailed down the design.
How deep can we go with relationships in a first version? Making the connections between proteins, structures, and compounds visible and navigable across three databases is the core value proposition. It’s also the hardest part. The shared UniProt ID gets us surprisingly far, but some types of relationships (like protein-to-protein interactions) might require additional data sources like STRING. How much we can do with just three databases is still an open question.
How do we keep up with change? These databases aren’t frozen. They add new entries, revise existing ones, change their data formats, and update their APIs. Any layer we build has to track those changes automatically or it goes stale fast. We need to design for this from day one.
Is three databases enough to convince anyone? Drug discovery uses far more than three databases. Genomic data, clinical variants, pathway maps, gene expression data, they all come into play at different stages. We think UniProt, ChEMBL, and PDB are enough to prove the concept for early-stage Discovery workflows, and we’ve already identified candidates for a second round of expansion. But whether three is convincing enough to build a business around remains a hypothesis.
These questions will determine whether this is a real startup or just an interesting idea that doesn’t survive implementation.
Three reasons.
The timing works. New AI systems for biology are appearing constantly. Every one of them needs data, and nearly every team we’ve talked to describes some version of the same integration pain. The customer base for this infrastructure is growing, but the infrastructure itself barely exists.
It’s hard to copy. Understanding biological data well enough to normalize, structure, and connect it requires real domain expertise. Each database you add makes the system more valuable. Each format change you handle builds knowledge that’s hard to replicate. The work compounds, which means being early matters.
It matters beyond business. Drug discovery is slow, expensive, and fails most of the time. If better data infrastructure makes AI even slightly more effective at the Discovery stage, the downstream effects on cost, speed, and patient outcomes could be meaningful. That’s not why we’d build it (we’d build it because it’s a good business problem), but it doesn’t hurt to be working on something that matters.
We’re in research mode. We’re looking closely at the raw data: how UniProt entries are structured at the field level, how PDB organizes its structure files, how ChEMBL records compound-target relationships. We need to understand the data deeply before we can design the right abstraction on top of it.
At the same time, we’re talking to potential users. Bio-AI startups, pharma data teams, bioinformatics researchers. Testing whether the pain points we’ve identified match what they actually deal with.
We’ll keep sharing what we find. This is a build-in-public project, and we think the fastest way to sharpen a hard idea is to put it in front of people early.
If you’re in this space, we’d love to hear from you. Are we seeing the problem right? What are we missing? Where are we wrong?
There are 2,236 active molecular biology databases in the world as of January 2025, according to the Nucleic…
The model wars are still on. But this week, the real battleground shifted to deployment. June 1, 2026…
Weekly Briefing The Big Four bet 750,000 seats on Claude. A Fields Medalist says GPT did PhD-level math…